

 产业资讯
产业资讯
 2017-11-27
2017-11-27
 4100
4100
来源:国际药政通 2017-11-27
“欧洲药品管理局(European Medicines Agency,EMA)在2017年11月22日出台了EudraVigilance的改良版,EudraVigilane是药物疑似不良反应的信息系统,包括已经在欧洲经济区(European Economic Area,EEA)内获批或正在进行临床试验研究的药物。新的系统使上市许可持有人和临床试验发起人更容易报告疑似不良反应,并可以更好地分析这些信息,从而为欧洲患者的安全带来益处。
根据Regulation (EC) No 726/2004第3款,EMA与成员国和欧盟委员会共同制定EudraVigilance数据库的功能规范。在2017年5月3日通过的建议中,已经确认EudraVigilance数据库具有完善的功能且系统满足功能规范要求。”
下列法律条款规定适用于2017年11月22日后通过EudraVigilance系统递交的强制性电子报告,法律条款如下:
•指令2001/83/EC第IX章“药物警戒”第3节中“药物警戒数据的记录、报告和评估”的第1条“疑似不良反应的记录和报告”;
•法规(EC)726 / 2004第II章“药物警戒”第3节中“人用药品的授权与监督”的第24(4)、28(1)、28a(1)(c)和28c(1)条。
各成员国应当确保上市许可持有人以电子方式向EudraVigilance数据库提交关于疑似不良反应信息的责任,由国家主管部门和上市许可持有人向EudraVigilance提供疑似不良反应的简化电子报告。
最新的欧盟药物警戒法律对疑似不良反应电子报告的要求进行了重大修改,以支持更完善的药品安全监测体系,为利益相关者提供更有效的系统。
当前对利益相关者的要求
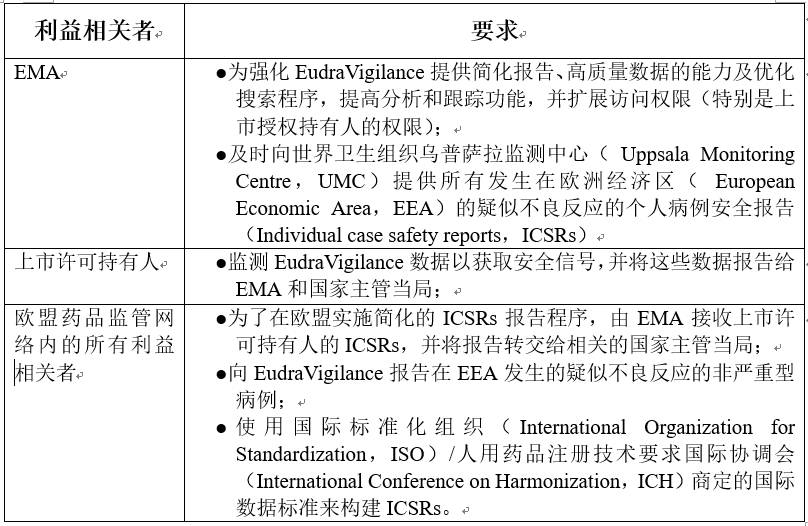
EudraVigilance系统的新功能和优势
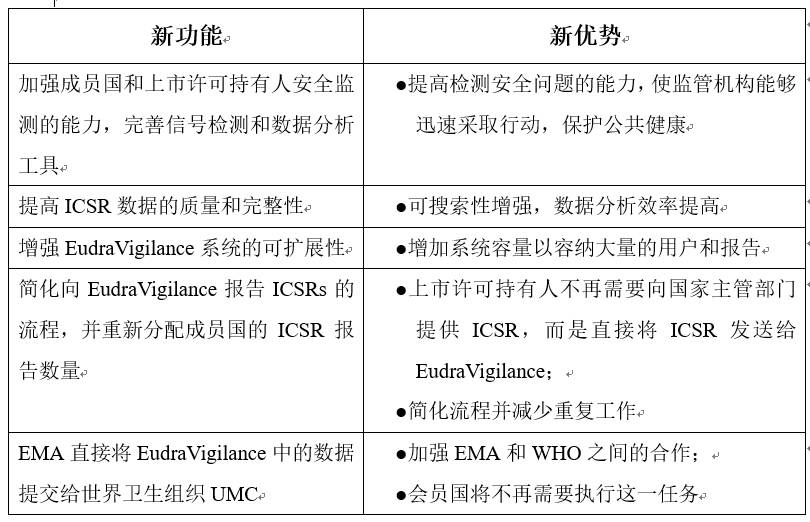
根据当地自发报告系统,由患者和医务人员向国家主管部门报告不良反应的规则将保持不变。在临床试验中,除非实行新的临床试验规范,否则临床试验期间的疑似严重不良反应报告规则也不会发生任何改变。
文章来源
http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2017/11/news_detail_002858.jsp&mid=WC0b01ac058004d5c1
 产业资讯
产业资讯
 医麦客 2025-03-12
30
医麦客 2025-03-12
30
 产业资讯
医药魔方Info 2025-03-12
28
产业资讯
医药魔方Info 2025-03-12
28
 产业资讯
医药观澜 2025-03-12
29
产业资讯
医药观澜 2025-03-12
29
 微信公众号
微信公众号 热门资讯
热门资讯 热点标签
热点标签