

 产业资讯
产业资讯
 识林
识林  2026-01-19
2026-01-19
 11
11
近日,ICH在其指南列表页面悄无声息地更新了一个条目,即“M16 EWG 结构化产品质量申报”(暂译,Structured Product Quality submissions,SPQS)。ICH组建了以FDA CDER药品质量办公室(OPQ)药品质量审评第1办公室(OPQA I)主任Geoffrey Wu为报告人的专家工作组,开始编制M16的概念性文件。
相比2025年5月ICH发布全新CTD架构M4Q(R2)征求意见引发广泛关注,SPQS尚不为业界所熟知。但早在ICH M4Q(R2)草拟阶段,ICH就已明确将在前者开始征求意见时启动SPQS。
具体何为SPQS,将给药学注册审评带来何种革新,这些疑问有待ICH在M16概念性文件和指南中阐述,但仅从其字面“结构化产品质量申报”理解,SPQS的内涵和外延可能比许多业界人士预期的要更深更广,可能超出M4Q(R2)重组药学申报资料的范畴。
其原因在于,由FDA人士主导的ICH SPQS可能延续了该机构近年来在药学(涵盖FDA语境下的Quality和CMC)注册审评领域的未竟创新,并有机会与日新月异的AI技术结合,充分发挥数据潜能。
注册和审评是一个硬币的两面。药业关注审评,也是在关注即将到来的注册范式变革。笔者基于FDA质量审评核心团队(包括余煊强博士)发表于2024年的综述文章《旨在改进 FDA 对人用药申请质量评估的监管创新网络》,将FDA的药学审评创新工具概述如下,供读者参考。
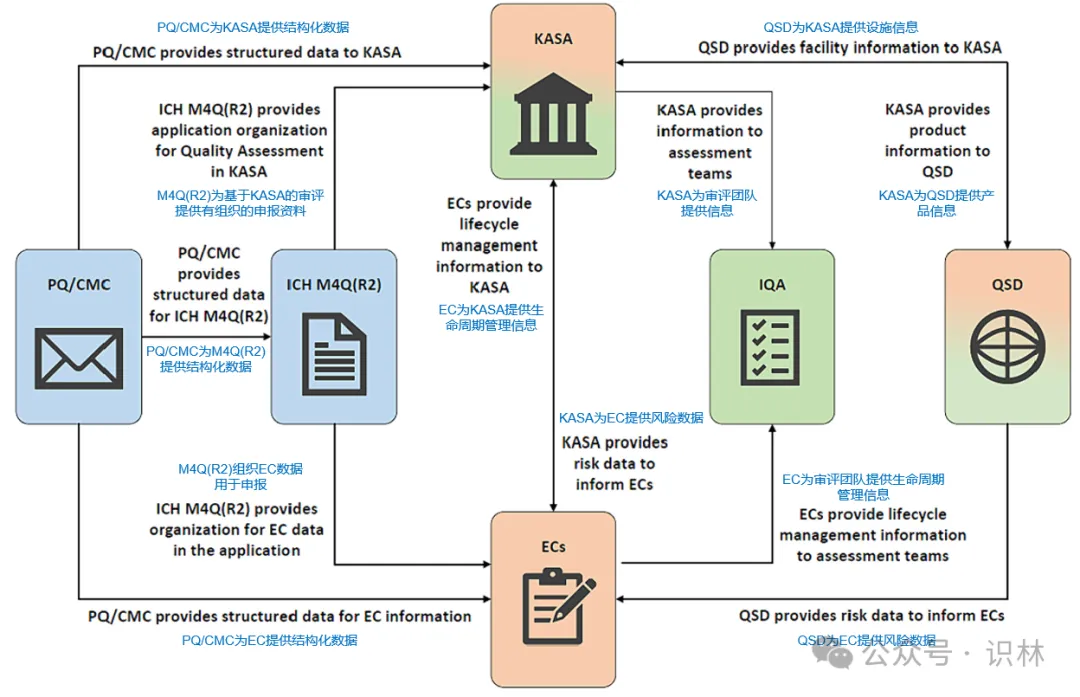
*FDA的药学审评创新工具矩阵,基于文献内图片编译
KASA:从“看资料”转向“读数据”
知识辅助评价和结构化申请(Knowledge-aided Assessment and Structured Application, KASA)是FDA首批监管创新之一。该系统利用信息技术,通过结构化数据、分析和知识管理来精简监管申报、审评和注册流程。
以往的药学审评方法依赖于审评人员对申请人提供的PDF文件信息进行自由叙述式(free narrative)描述(申报资料本身也是叙述式)。此类系统可能导致文本冗长、知识共享能力不一致且受限、数据提取困难,影响决策效率。KASA系统以结构化格式捕获并管理关于原料药、制剂、生产及设施的风险与控制策略信息,通过为各审评学科建立规则和算法来辅助审评人员,以促进风险分析、缓解和沟通。风险识别涉及计算机辅助分析申请材料是否符合监管标准,并基于先前知识确定产品在整个生命周期内的质量风险。KASA还结构化审评文件,以最小化不必要的文本叙述以及对申请人已提供信息的重复总结。
读者可点击此文更直观了解KASA。
PQ/CMC:“读数据”的前提是数据结构和交换标准
尽管ICH M4Q(R2)提供了有利于基于科学和风险的药学审评的组织结构,但它未涉及数据结构和交换标准。缺乏这些标准,无论CTD目录如何改动,最终提交的仍可能是pdf而不是数据。
药品质量/化学、生产与控制结构化数据(Structured Data on Pharmaceutical Quality/Chemistry, Manufacturing, and Controls, PQ/CMC)工作的重点就是建立此类标准,以实现从提交申报资料到监管机构审评数据的无缝连接和传输,可供KASA、质量监控仪表盘(Quality Surveillance Dashboard, QSD)及其他FDA系统使用。结构化数据的益处在于确保FDA和行业使用“相同的数据”,并消除在分析系统使用前手动操作数据的需要。
EC:结构化数据有望助Q12重要工具发扬光大
既定条件(Established Conditions, EC)的概念首见于FDA 在2015年5月发布《既定条件:已批准的药品和生物制品的可报告的化学、生产、控制(CMC)变更》指南草案。ICH 于2021年5月定稿Q12之后,EC作为Q12三大监管工具之一进入我国业界视野。另外两个工具是批准后变更管理方案(PACMP)和产品生命周期管理(PLCM)。
EC被定义为药品申请中那些为保证产品质量所必需的CMC要素(例如,生产工艺、设施和设备以及相关控制策略的要素)。FDA原本期望利用EC减少补充申请审评压力。然而在实践中,监管和业界双方对于何为EC难以达成共识,而对于EC的僵化理解可能对变更管理产生负面影响,导致企业“该报不报”或是“冗余申报”,以至于打击企业推动优化质量的变更的积极性。近日日本业界的EC调研就显示药企对于EC应用严重不足。
值得一提的是,我国药监已发布了多份PACMP指南,但对EC还在评估当中。NMPA在2025年12月的Q12适用问题公告中表示“将持续跟进国际进展,及时借鉴国际经验,组织工业界开展研究,积极推动...转化应用”。
在FDA看来,EC想要充分发挥价值,需要整个结构化申报体系的支持,如KASA将捕获产品整个生命周期中EC的风险评估,而QSD将促进知识管理及设施评估,帮助FDA评价企业有效管理变更的能力。
QSD:“仪表盘”链接监管内部“数据孤岛”
设施审评需要对产品属性、生产工艺和检查历史进行全面评价。然而相关数据分布在FDA内部多个不同的数据系统中。药学审评人员可能花费大量时间手动检查、提取和审评每个系统的数据。执行高效的设施审评需要一个集中化和自动化的系统,提供全面且最新的设施质量相关信息。
质量监控仪表盘(QSD)的设计初衷正是在一个平台汇总多源设施信息进行报告、数据探索和分析,为CDER提供一致审评的框架。该仪表盘为审评人员提供导航每个设施产品详细信息(例如,申请状态、供应链角色、给药途径、产品可及性)以及设施历史事件时间线的能力。它同时使用预测模型和自然语言处理(检测和提取文本数据关系的自动化技术),以实现高效的基于风险的设施审评。未来版本的QSD将集成从KASA收集的数据,获得更强大的分析能力。随着QSD的成熟,它将扩大用户数量,扩展数据获取范围,并改进数据清洗方法。不过FDA特别强调,执行审评的仍然是审评人员而非QSD。
IQA:基于强大数据基础的审评团队建设和工作流程设计
FDA认为,审评创新并非一定是电子化和数字化,高效药学审评总是需要不同质量学科的无缝整合。集成质量审评(Integrated Quality Assessment, IQA)流程促进了一种多学科、基于团队的方法,遵循明确的业务流程,并为每个团队成员设定清晰的职责。IQA团队包括申请技术负责人、监管业务流程经理、学科审评人员以及根据需要增加的技术顾问。
IQA面临的挑战之一是需要团队建设和关系建设,即团队内部的凝聚力和一致性,并需要精心设计以将不同学科的个体聚集在一起,有效处理大量申请。药品质量办公室(OPQ)在设计IQA团队时采用“对齐团队”策略。该策略从跨学科的较小人员池中抽取人员分配对齐的IQA团队,从而形成更频繁的合作,因此拥有更稳固的工作关系和增强信任的审评人员小组。这使得团队能够尽可能协作且高效地进行药学审评,同时为申请人提供更一致的反馈。
OPQ于2015年首次采用IQA流程,其积极经验还用于支持新药办公室(OND)在2020年实施集成新药申请(NDA)审评团队。未来OPQ将继续监控对齐团队以寻找持续改进的机会。
原文链接:
https://www.ich.org/page/multidisciplinary-guidelines
 产业资讯
识林 2026-01-19
11
产业资讯
识林 2026-01-19
11
 产业资讯
识林 2026-01-19
12
产业资讯
识林 2026-01-19
12
 产业资讯
生物制药小编 2026-01-19
12
产业资讯
生物制药小编 2026-01-19
12
 微信公众号
微信公众号 热门资讯
热门资讯 热点标签
热点标签